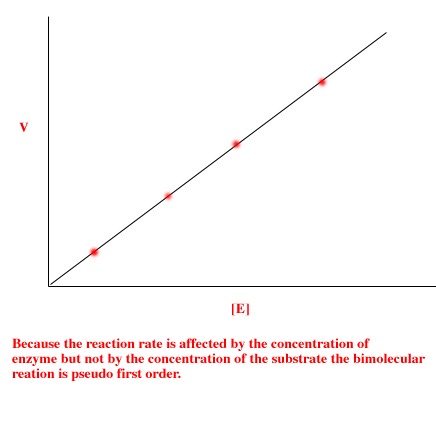
|
Phase Contrast Microscopy
Most of the detail of living cells is undetectable in bright field microscopy because there is too little contrast between structures with similar transparency and no color. Unless the specimen mount is extremely thin, dark field mode may distort details. However, the various organelles show wide variation in refractive index, that is, the tendency of the materials to bend light, providing an opportunity to distinguish them. Highly refractive structures bend light to a much greater angle than do structures of low refractive index. The same properties that cause the light to bend also delay the passage of light by a quarter of a wavelength or so. In a light microscope in bright field mode, light from highly refractive structures bends farther away from the center of the lens than light from less refractive structures and arrives about a quarter of a wavelength out of phase. Light from most objects passes through the center of the lens as well as to the periphery. If the light from an object to the edges of the objective lens is retarded a half wavelength and the light to the center is not retarded at all, then the light rays are out of phase by a half wavelength. They cancel each other when the objective lens brings the image into focus. A reduction in brightness of the object is observed. The degree of reduction in brightness depends on the refractive index of the object. Phase Contrast Condensers and Objective Lenses To use phase contrast the light path must be aligned. An element in the condenser is aligned with an element in a specialized phase contrast lens. This usually involves sliding a component into the light path or rotating a condenser turret. The elements are either lined up in a fixed position or are adjusted by the observer until the phase effect is optimized. Generally, more light is needed for phase contrast than for corresponding bright field viewing, since the technique is based on a diminishment of brightness of most objects. |
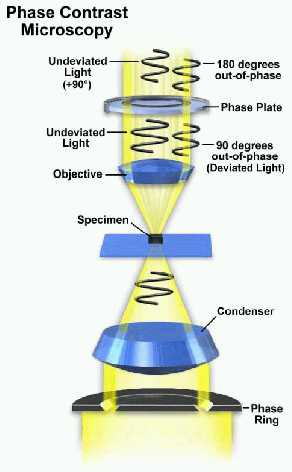 Schematic configuration for phase contrast microscopy: Light passing through the phase ring is first concentrated onto the specimen by the condenser. Undeviated light enters the objective and is advanced by the phase plate before interference at the rear focal plane of the objective.
Schematic configuration for phase contrast microscopy: Light passing through the phase ring is first concentrated onto the specimen by the condenser. Undeviated light enters the objective and is advanced by the phase plate before interference at the rear focal plane of the objective.
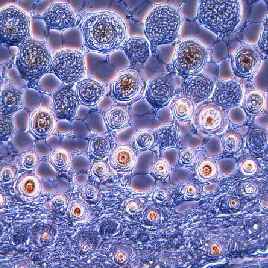 Photomicrograph of hair cross sections from a fetal mouse using phase contrast microscopy (200X).
Photomicrograph of hair cross sections from a fetal mouse using phase contrast microscopy (200X).
|
|
Polarized Light Microscopy
Many transparent solids are optically isotropic, meaning that the index of refraction is equal in all directions throughout the crystalline lattice. Examples of isotropic solids are glass, sodium chloride, many polymers, and a wide variety of both organic and inorganic compounds. Crystals are classified as being either isotropic or anisotropic depending upon their optical behavior and whether or not their crystallographic axes are equivalent. All isotropic crystals have equivalent axes that interact with light in a similar manner, regardless of the crystal orientation with respect to incident light waves. Light entering an isotropic crystal is refracted at a constant angle and passes through the crystal at a single velocity without being polarized by interaction with the electronic components of the crystalline lattice. Anisotropic crystals have crystallographically distinct axes and interact with light in a manner that is dependent upon the orientation of the crystalline lattice with respect to incident light. When light enters non-equivalent axes, it is refracted into two rays, each polarized with their vibration directions oriented at right angles to each other and traveling at different velocities. One of the rays travels with the same velocity in every direction through the crystal and is termed the 'ordinary ray'. The other ray travels with a velicity that is dependent upon the propagation direction within the crystal and is termed the 'extraordinary ray'. A polarizer is placed beneath the substage condenser causing polarized light to enter the anisotropic crystal where it is refracted into two separate component rays vibrating parallel to the crystallographic axes and perpendicular to each other. The polarized light waves then pass through the specimen and objective lense before reaching a second polarizer, termed the 'analyzer', that is oriented to pass a polarized vibration direction perpendicular to that of the substage polarizer. |
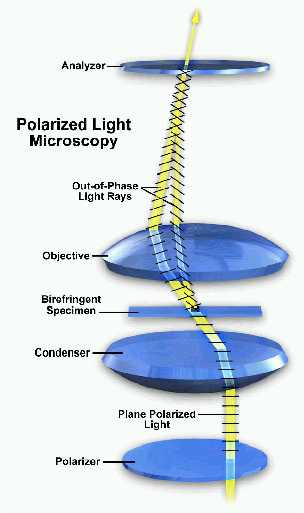 Schematic configuration for polarized light microscopy: White light is plane polarized by the substage polarizer and concentrated onto the anisotropic specimen by the condenser. Light rays emerging from the specimen interfere when they are recombined in the analyzer, subtracting some of the wavelengths of white light and producing a myriad of colors.
Schematic configuration for polarized light microscopy: White light is plane polarized by the substage polarizer and concentrated onto the anisotropic specimen by the condenser. Light rays emerging from the specimen interfere when they are recombined in the analyzer, subtracting some of the wavelengths of white light and producing a myriad of colors.

Photomicrograph of high-density liquid crystalline calf thymus DNA using polarized light microscopy (100X). |
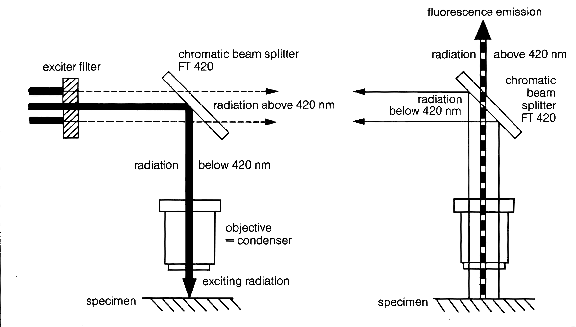
| Fluorescence of a substance is seen when the molecule is exposed to a specific wavelength of light (excitation wavelength or spectrum) and the light it emits (the emission wavelength or spectrum) is always of a higher wavelength. To view this fluorescence in the microscope, several light filtering components are needed. Specific filters are needed to isolate the |
|
| One other component is required: a dichroic beam splitter or partial mirror which reflects lower wavelengths of light and allows higher wavelengths to pass. A beam splitter is required because the objective acts as a condenser lens for the excitation wavelength as well as the objective lens for emission. One only wishes to see the light emitted from the fluorochrome and not any of the excitation light, and the beam splitter isolates the emitted light from the excitation |
|
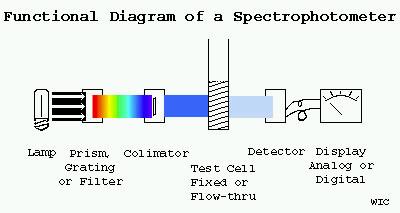 Diagram of the basic components of a spectrophotometer, consisting of light source (Lamp), Prism or grating, Colimator, Cuvette (Test Cell), Detector (selenium or silicon photocell or photomultiplier tube) and Display (analog or digital). In some implementations the Lamp, Prism and Colimator are a separate unit known as a 'Monochromator'. Physical arangements and enhancements vary with specific applications.
Diagram of the basic components of a spectrophotometer, consisting of light source (Lamp), Prism or grating, Colimator, Cuvette (Test Cell), Detector (selenium or silicon photocell or photomultiplier tube) and Display (analog or digital). In some implementations the Lamp, Prism and Colimator are a separate unit known as a 'Monochromator'. Physical arangements and enhancements vary with specific applications.
|
 Spectronic 20 Spectrophotometer: It is manufactured in several configurations.
Spectronic 20 Spectrophotometer: It is manufactured in several configurations.
|
where,
I0 = incident radiation, I = transmitted radiation
A = absorbance
e = extinction coefficient at a given wavelength
l = pathlength in cm.
C = molar concentration, [M]
(The extinction coefficient, molar absorptivity, has units M-1cm-1; hence, most standard cuvette cells have a pathlength of 1 cm.)
Cu = 0.622/[(6.22 mM-1cm-1)(1 cm)] = 0.1 mM NADH
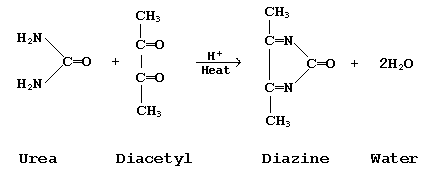 In the Fearon procedure for serum urea quantitation, urea condenses with diacetyl to form diazine. Since diacetyl is unstable, diacetyl monoxime is substituted and generates the required diacetyl in the same reaction mixture. Also, thiosemicarbazide and ferric ions are added to enhance and stabalize the product.
In the Fearon procedure for serum urea quantitation, urea condenses with diacetyl to form diazine. Since diacetyl is unstable, diacetyl monoxime is substituted and generates the required diacetyl in the same reaction mixture. Also, thiosemicarbazide and ferric ions are added to enhance and stabalize the product.
|
virtually transparent to all practical visible and uv wavelengths, the new product, diazine, absorbs strongly at 540 nm and its concentration is directly related to the concentration of urea.
Calibration Calibration of a method is applied using a wide variety of mathematical methods or mathematical models. In practice, nearly all photometric methods require calibration with standard solutions of known value because of |
where Cu = Concentration of the Unknown solution
Cs = Concentration of the Standard solution
Au = Absorbance of the Unknown solution
As = Absorbance of the Standard solution
INFRARED SPECTROSCOPY
|
IR is an absorption form of spectrometric analysis. Unlike atomic absorption, IR is not concerned with specific elements (such as Lead, Copper, etc.) but, rather, with the groupings of atoms in specific combinations to form what are called "functional groups". These various functional groups help to determine a material's properties or expected behavior.
By knowing which wavelengths are absorbed by each functional group of interest one can cause the appropriate wavelength to be directed at the sample |
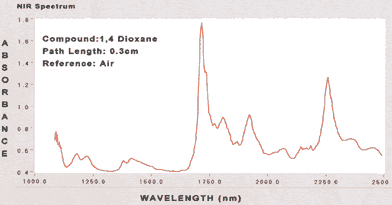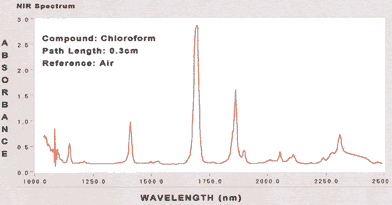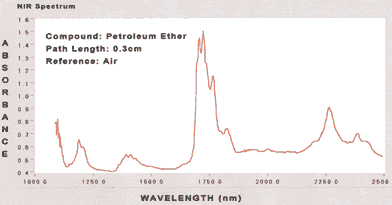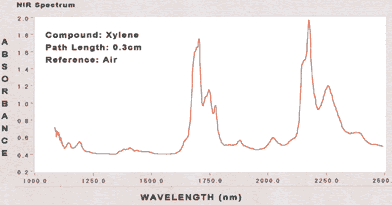
|
Small spot size (10-15 microns)
Detailed chemical bonding information
Organic analysis of polymers and plastics
Analysis of liquids, solids, and gases
In ATR mode can sample the outer ~ 1000 �
Non-destructive analysis
Can analyze non-conductive materials
Molecular specific identification
 yi x yj dv >> 0]. A large Raman signal requires a large change in the polarizability of the molecule [aij = yi ayj dv >>0 ]. While many transitions have both large mij and large aij, there are many that don't. Therefore, Raman and IR can act together to give a more complete description of the molecule.
yi x yj dv >> 0]. A large Raman signal requires a large change in the polarizability of the molecule [aij = yi ayj dv >>0 ]. While many transitions have both large mij and large aij, there are many that don't. Therefore, Raman and IR can act together to give a more complete description of the molecule.
 In Gas Chromatography, the determining factor in how fast a component travels is usually (but not always) the boiling point of the compound. (If a polar high-boiling liquid adsorbent is used in the GC column, the polarity of the components determines the elution order.)
In Gas Chromatography, the determining factor in how fast a component travels is usually (but not always) the boiling point of the compound. (If a polar high-boiling liquid adsorbent is used in the GC column, the polarity of the components determines the elution order.)
|
Gas chromatography is an important method owing to its speed, resolving power, and detector sensitivity. Since it depends on vaporization, this technique is best suited to compounds that can be vaporized without suffering decomposition. Many substances that normally do not easily vaporize can be chemically derivatized for successful volatilization separation by gas chromatography.
Right-click and click Reload to view the animation at left. |
|
Leaving the entire capillary GC system aside, the major components of the mass selective detector itself are: an ionization source, mass separator, and ion detector. There are two common mass analyzers or separators commercially available for GC/MS; they are the quadrapole and the ion trap.
Right-click and click Reload to view the animation at right. In this example, the lightest fragment is B+; the heaviest A+. The last frame of the movie is a mass spectrum displaying only these three fragments. Their relative mass to charge ratios are specified by their relative position on the x axis (low mass/charge to left, high mass/charge to right). The relative amounts (commonly called peak intensity) of each of these fragments determined during the mass analyzer's scan is reflected on the y axis. GC/MS is a reference method used in the clinical laboratory to detect, identify and quantitate DAU (Drugs of AbUse) and by forensic toxicology laboratories. Click here for example IR and Mass spectra (Cocaine). |
 The GIF animation is a short series of steps for the process of a single analyte (already separated from the other analytes in the chromatographic mixture) denoted as ABC exiting the chromatographic column and: the analyte (A-B-C) undergoing ionization and fragmentation; the charged fragments (A+ B+ C+) being separated by mass; the fragments which are focused on the mass filter's exit slit passing into the detector; and the charged ions being detected.
The GIF animation is a short series of steps for the process of a single analyte (already separated from the other analytes in the chromatographic mixture) denoted as ABC exiting the chromatographic column and: the analyte (A-B-C) undergoing ionization and fragmentation; the charged fragments (A+ B+ C+) being separated by mass; the fragments which are focused on the mass filter's exit slit passing into the detector; and the charged ions being detected.
|
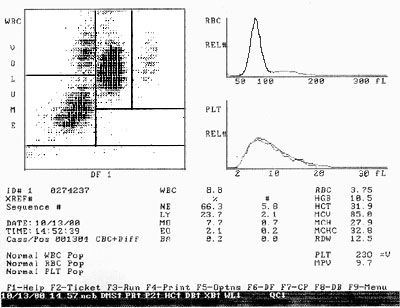
|
Flow cytometers have been commercially available since the early 1970's, and their use has been increasing since then. The most numerous flow cytometers are those used for complete blood cell counts in clinical laboratories -- these do not employ fluorescence. More versatile research instruments employ fluorescence, hence may be distinguished as flow cytofluorometers.
< Screen print of Coulter MAXM hemogram. Cell types are classified by size, volume and internal structure. |
low angle forward scatter intensity, approximately proportional to cell diameter
orthogonal (90 degree) scatter intensity, approximately proportional to the quantity
of granular structures within the cell
fluorescence intensities at several wavelengths
Light scatter alone is often quite useful. It is commonly used to exclude dead cells, cell aggregates, and cell debris from the fluorescence data. It is sufficient to distinguish lymphocytes from monocytes from granulocytes in blood leukocyte samples.
Enzymes are extraordinarily efficient and selective biological catalysts that accelerate the chemical reaction toward equilibrium. Chemical reactions in the metabolic pathways necessary for the maintenance of life would not proceed at reasonable rates without enzymes. Enzymatic reactions are 103 to 1017 times faster than the corresponding uncatalyzed reactions.
Enzymes are globular protein molecules (M.Wt. typically 104 - 105 ) that catalyze the myriad of biochemical reactions that occur within living cells. Like their chemical counterparts, enzymes accelerate the rate of chemical reactions without themselves being changed in the overall process. A fundamental difference between enzymes and industrial catalysts however is that enzymes function at physiological temperatures in a low ionic strength solution at near neutral pH. There are many different kinds on enzyme, each promoting only a very limited range of chemical reactions. Enzymes are very efficient catalysts. One molecule of catalase for example can decompose 40,000 molecules of hydrogen peroxide per second at freezing point.
The table below gives turnover numbers for some other enzymes at room temperature.
Enzyme Turnover number
(molecules/second)
Carbonic anhydrase 600,000
Acetylcholinesterase 25,000
amylase 18,000
Penicillinase 2,000
DNA polymerase 15
The substrate molecules are bound in a hydrophobic cleft know as the active site. Enzymes whose activity is regulated generally have a more complex structure than unregulated enzymes. Most regulatory enzymes have two sites - one for the substrate, and the other for the modulator.
Enzyme Kinetics
An important feature of enzymes is that they possess specific 3-D configurations that are fundamental to their biological function. This is because the overall shape of the molecule stabilizes the precise geometric structure of the active site, the region in the enzyme where the substrate is converted into the product. The importance of the active site (which normally makes up only a small percentage of the entire molecule) is to stabilize the transition state between the substrate and its products thus lowering the activation energy for the reaction. (Lowering this energy by about 34 kJ mol-1 is calculated as bringing a million fold increase in the rate of a reaction at 298 K.) For this to occur the substrate must fit precisely into the active site (shape recognition). Indeed, the "lock and key" analogy is often used to describe substrate binding, as substrate fits an enzyme like a key fits a lock. The activation energy for the enzyme catalyzed reaction can be determined in the normal way by the Arrhenius equation;
where k is the rate constant, Ze is a factor accounting for the frequency of collisions, R is the Gas constant, T is the absolute temperature and Ea is the activation energy. The activation energy may be determined by measurement of the reaction rate at different temperatures (limited to the thermal stability range of the enzyme) and plotting ln(k) against 1/T.
Each enzyme requires certain definite conditions for optimum performance, particularly as regards to pH, temperature and ionic strength. The presence of specific accessory substances (co-factors, activators etc.) may also be a requirement.
Enzymes are unstable substances, and are easily inactivated by high temperature or extremes of pH. Although they are not consumed during the biochemical reaction, enzymes are inherently labile and have to be continuously synthesized.
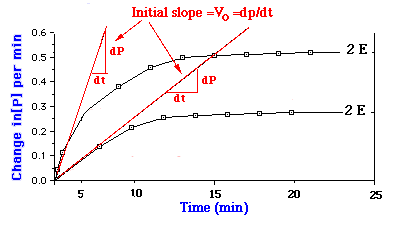
For many enzyme reactions the rate (V or dp/dt) varies with the substrate concentration [S] as shown below. The rate (V or dp/dt) is defined as the number of moles per second which is a measure of enzyme activity. At low substrate concentration the rate or velocity (V) is almost proportional to substrate[S] concentration. At high substrate [S] concentrations, the velocity is not linear with the [S] concentration and rate approaches a maximum velocity called Vmax.
The following model was proposed by Michaelis and Menten in 1913 to explain
the kinetics of an enzyme reaction (equation 1). One of first great advances
in biochemistry was the discovery that an enzyme transiently binds to a
substrate to form a enzyme-substrate complex [ES]. The substrate binds noncovalently
to the active site of the enzyme. The rate of an enzymatic reaction depends
on the concentrations of both the substrate and the catalyst (enzyme).
When the amount of enzyme is much less than the amount of substrate, the
reaction is pseudo first order. The straight line illustrates the effect
of enzyme concentration on reaction velocity in pseudo first-order reaction.
The more enzyme present, the faster the reaction.
|
|
Pseudo first-order conditions are used in analyses that determine the
concentration of an enzyme in a sample. The concentration of enzyme can
be easily determined by comparing its activity to a reference curve as
shown at left.
Modern automated clinical chemistry analyzers store the kinetic reaction constants in their computer and calculate unknown values from measured absorbance changes over standard time. This implementation of enzyme measurement is known as a kinetic method. In fact, most analyses (not merely for enzymes) employ a kinetic method - because they are faster. End-point methods often require up to an hour or more for the reaction to achieve equilibrium, whereas kinetic methods may require just a few minutes. |
At the beginning of an enzyme-catalyzed reaction, the amount of product
formed is negligible, and the reaction can be described by equation 1.
Note that the conversion of the ES complex into free enzyme and product
is shown by a one-way arrow. During the initial period when measurements
are made little product has been formed, so the rate of the reverse reaction
is small. The velocity measured during this short period is called the
initial velocity (vo). The use of initial velocity simplifies
the interpretation of kinetic data and avoids complications that may arise
as the reaction progresses, such as product inhibition, depletion of substrate,
and slow denaturation of the enzyme.
Equation 1:
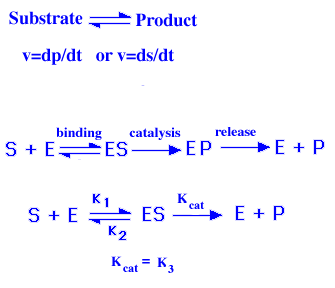
An enzyme (E) combines with [S] to form an ES complex, with a rate constant k1. The ES complex has two possible fates. It can dissociate to form E and S, with a rate constant k2, or it can proceed to form product P, with a rate constant k3 (kcat). It is assumed that almost none of the product reverts to the initial substrate. The velocity of the reaction is then V (dp/dt) is represented by equation 2.
Equation 2:
Expressing [ES] in terms of known quantities, the rates of formation and breakdown of ES are given by:
Equation 3:
Equation 4:
We are interested in the catalytic rate under steady-state conditions. With steady-state the concentration of the intermediate [ES] stays the same while the concentrations of the starting material and product are changing. Equation 5 represents steady-state conditions.
Equation 5:
Rearranging equation 5,
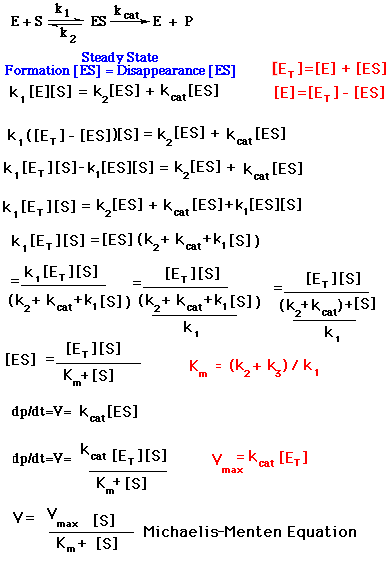
The maximal rate, Vmax, is attained when the enzyme sites are saturated with substrate. The maximal velocity is when [S] is much greater than the value of Km so that [S]/([S] + Km) approaches 1 as shown in equation 6. Thus,
Equation 6:
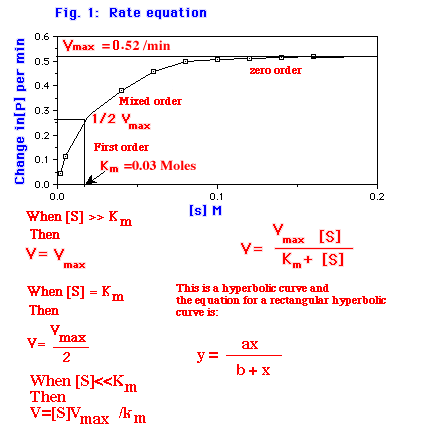
The Michaelis-Menten equation accounts for the kinetic data given in Figure 1. At very low substrate concentration, when [S] is much less than Km then the velocity is as shown in equation 7:
Equation 7:
In equation 7 the velocity is directly proportional to the substrate concentration. At high substrate concentration, when [S] is much greater than K m, the velocity (V) is the maximal rate (Vmax), and independent of the substrate concentration which depends on the total enzyme concentration, E, and rate constant k3 as shown by this in equation 6.
The meaning of Km is evident from the Michaelis-Menten equation. When [S] = Km, then V=Vmax/2. Thus, Km is equal to the substrate concentration at which the reaction rate is half of its maximal value.
The Michaelis constant, Km, and the maximal rate, Vmax, can be readily derived from rates of catalysis measured at different substrate concentrations if an enzyme operates according to the simple scheme given in the Michaelis Menten equation. It is convenient to transform the Michaelis-Menten equation into one that gives a straight line plot. This can be done by taking the reciprocal of both sides of the equation.
A plot of 1/V versus 1/[S], called a Lineweaver-Burk plot, yields a straight line with and intercept of 1/Vmaxand slope of Km/Vmax
Figure 2.
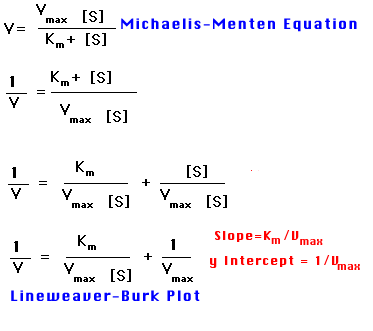
Table 1: Rate equation data
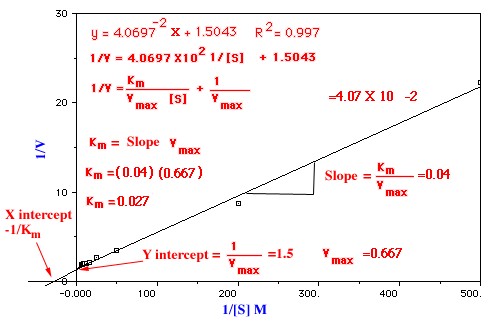
Figure 1: The plot is the reaction velocity V verses the concentration of the substrate [S]. The maximum velocity Vmax is the velocity where the enzyme is saturated with substrate[S] and 1/2 Vmax is that substrate [S] where the velocity 1/2 Vmax.
The Michaelis constant, Km, and the maximal rate, Vmax can be readily derived from rates of enzyme catalyzed reaction measured at different substrate concentrations. The enzyme must operate according to the simple scheme given in equation 1.
It is convenient to transform the Michaelis-Menten equation into one that gives a straight line plot. This can be done by taking the reciprocal of both sides of the equation. A double-reciprocal plot of enzyme kinetics: 1/V is plotted as a function of 1/[S]. The slope is Km/Vmax, the intercept on the vertical axis is 1/Vmax, and the intercept on the horizontal axis is -1/Km.
Km and Vmax for an enzyme-catalyzed reaction can be measured in several ways. Both values can be obtained by analysis of initial velocities at a series of substrate concentrations and a fixed concentration for the enzyme. In order to obtain reliable values for the kinetic constants, the [S] point must be spread out both below and above Km to produce a hyperbola. It is difficult to determine either Km or Vmax directly from a graph of initial velocity versus concentration because the curve approaches Vmax asymptotically. However, using a suitable computer program, accurate values can be determined by fitting the experimental results to the equation for the hyperbola.
The Michaelis-Menten equation can be rewritten in order to obtain values for Vmax and Km from straight lines on graphs. The most commonly used transformation is the double-reciprocal Lineweaver-Burk plot. Values of Km can be determined even when enzymes have not been purified, provided that only one enzyme in the impure preparation can catalyze the observed reaction.
Table 2 Km values of some enzyme
| Enzyme | Substrate | Km (mM) |
| Chymotrypsin | Acetyl-L-tryptophanamide | 5,000 |
| Lysozyme | Hexa-N-acetylglucosamine | 6 |
| b-Galactosidase | Lactose | 4,000 |
| Threonine deaminase | Threonine | 5,000 |
| Carbonic anhydrase | CO2 | 800 |
| Penicillianase | Benzylpenicillin | 50 |
| Pyruvate Carboxylase | Pyruvate
HCO3- ATP |
400
1,000 50 |
| Arginine-tRNA synthetase | Arginine
tRNA ATP |
3
0.4 300 |
At high substrate concentration, when [S] is much greater than Km, V=Vmax, i.e., the rate is maximal, independent of substrate concentration. When V=Vmax then [S]/([S] + Km) approaches 1. When [S] = Km then V = Vmax/2. Thus Km is equal to the substrate concentration at which the reaction rate is half of its maximal value. When the [S] is less than Km then the V=Vmax [S]/Km and the substrate concentration is proportional to the velocity.
The significance of Km and Vmax values
The K m values of enzymes range widely (0.4 µM for Arginine-tRNA synthetase to 5,000 µM for chymotrypsin). For most enzymes Km lies between 10-1 and 10-7 M. The Km value for am enzyme depends on the particular substrate and also on environmental conditions such as pH, temperature, and ionic strength. The Michaelis constant, Km has two meanings. First, Km is the concentration of substrate at which half the active sites are filled. Once the Km is known, the fraction of site filled at any substrate concentration can be calculated from the equation
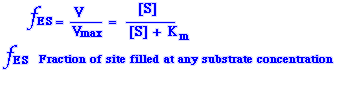
If you divide the velocity by maximal velocity then you get the number of sites filled fES =V/Vmax or if you use this equation fES = [S]/([S] + Km) then you get the fraction of site filled
Second K m is related to the rate constants of the individual steps in the catalytic scheme given in the following equation.
Km=(k2 + kcat)/k1
Equation 1:
Consider a limiting case in which k2 is much greater than kcat. This means that the dissociation of the ES complex to E and S is much more rapid than formation of E and Product. Under these conditions (k2 >>kcat) and Km equals.
K m =k2 /k1
The dissociation constant of the ES complex is given by
K ES =[E][S]/[ES]=k2 /k1
In other words, Km is equal to the dissociation constant of the ES complex if kcat is much smaller than k2. When this condition is met. Km is a measure of the strength of the ES complex: A high Km indicates weak binding ; a low Km indicates strong binding. It must be stressed that Km indicates the affinity of the ES complex only when k2 is much greater than kcat.
Turnover number
The turnover number of an enzyme is the number of substrate molecules converted into product by an enzyme molecule in a unit of time when the enzyme is fully saturated with substrate. It is equal to the kinetic constant kcat. The maximal rate Vmax, reveals the turnover number of an enzyme if the concentration of active site [ET] is known, because
V max =kcat [ET]
For example, a 10-6 M solution of carbonic anhydrase catalyzes the formation of 0.6 M H2CO3 per second when it is fully saturated with substrate. Hence, kcat is 6 X 105 s-1.
kcat= Vmax/Et = 0.6/10-6=6 X 105 S-1
This turnover number is one of the largest known. Each round of catalysis occurs in a time equal to 1/kcat, which is 1.7 microseconds for carbonic anhydrase. The turnover numbers of most enzymes with their physiological substrates fall in the range from 10 to 107 per second.
| Enzyme | Kcat(s-1) |
| Papain | 10 |
| Ribonuclease | 100 |
| Carboxypeptidase | 100 |
| Trypsin | 100 to 1,000 |
| Acetylcholinesterase | 1,000 |
| Kinases | 1,000 |
| Dehydrogenases | 1,000 |
| Transaminases | 1,000 |
| Carbonic anhydrase | 1,000,000 |
| Superoixe dismutase | 1,000,000 |
| catalase | 10,000,000 |
Kinetic Perfection in Enzymatic Catalysis (The K cat /K m)
When the substrate concentration is much greater than Km, the rate of catalysis is equal to k cat, the turnover number, as described in the preceding section. However, most enzymes are not normally saturated with substrate. Under physiological conditions, the [S]/Km ratio is typically between 0.01 and 1.0. when [S]<<Km, the enzymatic rate is much less than kcat because most of the active sites are unoccupied. Is there a number that characterizes the kinetic of an enzyme under these conditions? Indeed there is, as can be shown by combining two equations.

When [S]<<Km, the concentration of free enzyme, [E], is nearly equal to the total concentration of enzyme [ET], and so
V=kcat/Km([S][ET]
Thus, when [S} <<Km, the enzymatic velocity depends on the value of Kcat/Km and on [S]. Are there any physical limits on the value of kcat/Km? Note that this ratio depends on k1, k2, and kcat, as can be shown by substituting for Km:
kcat/Km=kcatk1/k2+kcat <k1
Thus the ultimate limit on the value of kcat/Km is set by k1, the rate of formation of the ES complex. This rate cannot be faster than the diffusion-controlled encounter of an enzyme and its substrate. Diffusion limits the value of k1 so that it cannot be higher than between 108 and 109 M-1S-1. Hence, the upper limit on kcat/Km is between 108 and 109 M-1S-1.
This restriction also pertains to enzymes having more complex reaction pathways. Their maximal catalytic rate when substrate is saturating, denoted by kcat, depends on several rate constants rather than on kcat alone. The pertinent parameter for these enzymes is kcat/Km. In fact, the kcat/Km ratios of the enzymes acetycholinesterase, carbonic anhydrase, and trisephophate isomerase are between 108 and 109 M-1 S-1, which shows that they have attained kinetic perfection. Their catalytic velocity is restricted only by the rate at which they encounter substrate in the solution. Any further gain in catalytic rate can come only by decreasing the time for diffusion. Indeed, some series of enzymes are associated into organized assemblies so that the product of one enzyme is very rapidly found by the next enzyme. In effect, products are channeled from one enzyme to the next, much as in an assembly line.
The constants kcat and kcat.Km are useful for comparing the activities of different enzymes. It is also possible to assess the efficiency of an enzyme by measuring the rate acceleration that it provides. This value is the ratio of the rate constant for a reaction in the presence of the enzyme (kcat) divided by the rate constant for the same reaction in the absence of enzyme(kn). Surprisingly few rate acceleration values are known because most cellular reactions occur extremely slowly in the absence of enzymes.
Competitive and Noncompetitive Inhibition
Measurements of the rates of catalysis at different concentrations of substrate and inhibitor serve to distinguish between competitive, uncompetitive and noncompetitive inhibition. An inhibitor (I) is a compound that binds to an enzyme and interferes with its activity by preventing either the formation of the ES complex or its breakdown to E +P. Inhibitors are used experimentally to investigate enzyme mechanisms and to decipher metabolic pathways. Natural inhibitors regulate metabolism, and many drugs are enzyme inhibitors. Inhibition can be either irreversible or reversible. Irreversible inhibitors are bound to enzymes by covalent bonds. Reversible inhibitors are bound to enzymes by the same noncovalent forces that bind substrates and products.
The constant for the dissociation of I from the EI complex, called the inhibition constant (Ki), is described by the equation Ki =[E][I]/[EI]
Effects of Reversible Inhibitors on Kinetic Constants
| Type of inhibitor | Effect |
| Competitive (I bind to E only) | Raises Km and Vmax remains unchanged |
| Uncompetitive (I binds to ES only | Lowers Vmax and Km Ratio of Vmax.Km
Remains unchanged |
| Noncompetitive (I binds to E or ES) | Lowers Vmax Km remains unchanged |
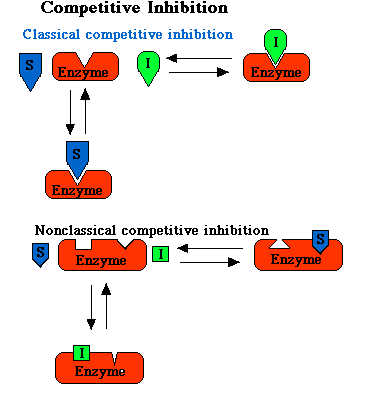
Competitive Inhibition
When a nonmetabolizable molecule, I, resembles a metabolizable molecule, S, sufficiently to be bound to the enzyme, then I remains attached to the enzyme and prevents the attachment of S, thus competing with S for space on the enzyme surface. The inhibition of the reactions of S is called classical competitive inhibition. Nonclassical competitive inhibition is the binding of S at the active site and prevents the binding of I at a different site. The reverse is also true.
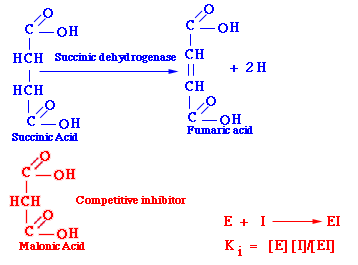
For example, malonic acid resembles succinic acid because both have two carboxyl groups. Malonic acid inhibits the action of succinic dehydrogenase on succinic acid by clogging the active site on the enzyme, and since malonic acid and succinic dehydrogenase does dissociate at a finite rate given by the dissociation constant. Therefore, an excess of succinic acid will reverse the action of malonic acid.
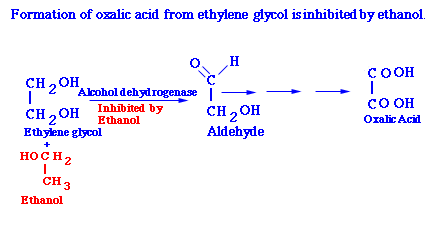
Ethanol is Used Therapeutically as a Competitive Inhibitor to Treat Ethylene Glycol Poisoning.
About fifty deaths occur annually from the ingestion of ethylene glycol, a constituent of antifreeze. Ethylene glycol itself is not lethally toxic. Rather, the harm is done by oxalic acid, the oxidation product of ethylene glycol, because the kidneys are severely damaged by the deposition of oxalate crystals. The first committed step in this conversion is the oxidation of ethylene glycol to an aldehyde by alcohol dehydrogenase. This reaction can be effectively inhibited by administering a nearly intoxicating dose of ethanol. The basis of this effect is that ethanol is a competing substrate and so it blocks the oxidation of ethylene glycol to aldehyde products. The ethylene glycol is then excreted harmlessly. Ethanol is also used as a competing substrate for treating methanol poisoning.
In competitive inhibition, the intercept of the plot of 1/V versus 1/[S] is the same in the presence and absence of inhibitor, although the slope is different. This reflects the fact that Vmax is not altered by a competitive inhibitor. Competitive inhibition can be overcome by a sufficiently high concentration of substrate. At a sufficiently high concentration, virtually all the active sites are filled by substrate and the enzyme is fully operative. The increase in the slope of the 1/v versus 1/[S] plot indicates the strength of binding of competitive inhibitor. In the presence of a competitive inhibitor the Lineweaver-Burk equation becomes:
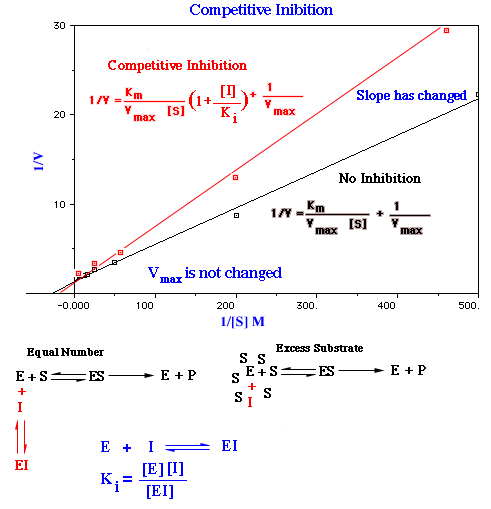
A double-reciprocal plot of enzyme kinetics in the presence and absence of a competitive inhibitor; Vmax is unaltered, whereas Km is increased.
In other words, the slope of the plot is increased by the factor (1+[I]/ki) in the presence of a competitive inhibitor. Consider an enzyme with a Km of 10-4M. In the absence of inhibitor, V=Vmax/2 when [S]=10-4 M. In the presence of 2 X 10-3 M competitive inhibitor that is bound to the enzyme with a Ki of 10-3 M, the apparent Km will be 3 X 10-4M. Substitution of these values into Lineweaver Burk equation give V=Vmax/4.
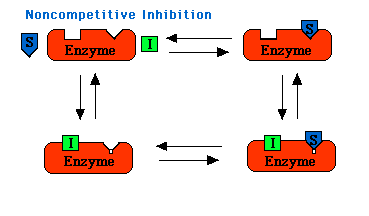
Noncompetitive Inhibition
In some cases the inhibitor resembles the metabolizable substance, but it is apparently attached to the enzyme at some point other than the one which binds the original substrate, and by its antagonistic action it prevents the expected enzyme reaction.
In noncompetitive inhibition Vmax is decreased to Vimax, and so the intercept on the vertical axis is increased. The new slope, which is equal to Km/VImax, is larger by the same factor. In contrast with Vmax, Km is not affected by this kind of inhibition. Noncompetitive inhibition cannot be overcome by increasing the substrate concentration. The maximal velocity in the presence of a noncompetitive inhibitor is Vimax.
Noncompetitive inhibitors probably alters the conformation of the enzyme to a shape that can still bind S but cannot catalyze any reaction by forming product.
.
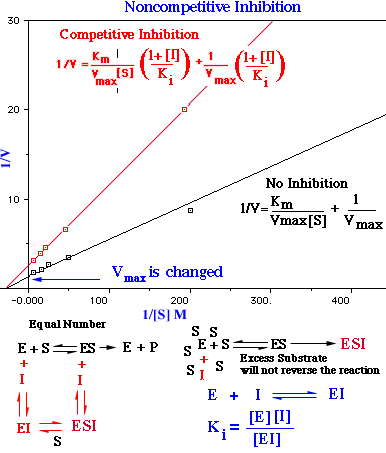
Uncompetitive Inhibition
Uncompetitive inhibitors bind only to ES, not to free enzyme. In uncompetitive inhibition, Vmax is decreased by the conversion of some molecules of E to the inactive form ESI. Since it is the ED complex that binds I, the decrease in Vit1 is not reversed by the addition of more substrate. Uncompetitive inhibitors also decrease the Km because the equilibria for the formation of both ES and ESI are shifted toward the complexes by the binding of I. Experimentally, the lines on a double-reciprocal plot representing varying concentrations of an uncompetitive inhibitor all have the same slope, indicating proportionally decreased value for Km and Vmax. This type of inhibition usually occurs only with multisubstrate reactions.
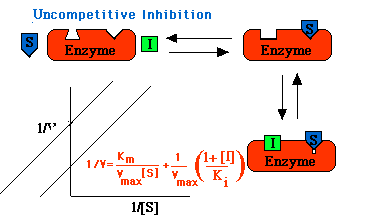
Classification of Enzymes
There are approximately 3000 enzymes which have been characterized. These are grouped into six main classes according to the type of reaction catalyzed. At present, only a limited number are used for analytical purposes.
Oxidoreductases
These enzymes catalyze oxidation and reduction reactions involving the transfer of hydrogen atoms or electrons. The following are of particular importance in the design of enzyme electrodes. This group can be further divided into 4 main classes.
Dehydrogenases catalyze hydrogen transfer from the substrate to a nicotinamide
adenine dinucleotide cofactor (NAD+). An example of this is lactate
dehydrogenase which catalyzes the following reaction:
Lactate + NAD+ = Pyruvate + NADH + H+
Oxidases catalyze hydrogen transfer from the substrate to molecular oxygen
producing hydrogen peroxide as a by-product. An example of this is FAD
dependent glucose oxidase which catalyses the following reaction:
b-D-glucose + O2 = gluconolactone + H2O2
Peroxidases catalyze oxidation of a substrate by hydrogen peroxide. An
example of this type of enzyme is horseradish peroxidase which catalyzes the
oxidation of a number of different reducing substances (dyes, amines,
hydroquinones etc.) and the concomitant reduction of hydrogen peroxide. The
reaction below illustrates the oxidation of neutral ferrocene to ferricinium in the
presence of hydrogen peroxide:
2[Fe(Cp)2] + H2O2 + 2H+= 2[Fe(Cp)2]+ + 2 H2O
Oxygenases catalyze substrate oxidation by molecular oxygen. The reduced
product of the reaction in this case is water and not hydrogen peroxide. An
example of this is the oxidation of lactate to acetate catalyzed by
lactate-2-monooxygenase.
lactate + O2 = acetate + CO2 + H2O
Transferases
These enzymes transfer C, N, P or S containing groups (alkyl, acyl, aldehyde, amino, phosphate or glucosyl) from one substrate to another. Transaminases, transketolases, transaldolases and transmethylases belong to this group.
Hydrolases
These enzymes catalyze cleavage reactions or the reverse fragment condensations. According to the type of bond cleaved, a distinction is made between peptidases, esterases, lipases, glycosidases, phosphatases and so on. Examples of this class of enzyme include; cholesterol esterase, alkaline phosphatase and glucoamylase.
Lyases
These enzymes non-hydrolytically remove groups from their substrates with the concomitant formation of double bonds or alternatively add new groups across double bonds.
Isomerases
These enzymes catalyse intramolecular rearrangements and are subdivided into;
racemases
epimerases
mutases
cis-trans-isomerases
An example of this class of enzyme is glucose isomerase which catalyses the
isomerisation of glucose to fructose.
Ligases
Ligases split C-C, C-O, C-N, C-S and C-halogen bonds without hydrolysis or oxidation. The reaction is usually accompanied by the consumption of a high energy compound such as ATP and other nucleoside triphosphates. An example of this type of enzyme is pyruvate carboxylase which catalyzes the following reaction:
An important aspect of catalytic action is the requirement by certain enzymes of either co-factors or prosthetic groups. Co-factors receive redox equivalents, protons or chemical groups from the substrate during the course of the enzymatic reaction. They tend to associate with the enzyme in a transient manner and can diffuse away from the active site. Examples of this type of molecule include NAD+ and NADP+.
Prosthetic groups have similar function to co-factors with the exception that they are tightly bound to the enzyme. When they are released, the enzyme is mostly denatured. Flavin nucleotides and hemes are the most important examples of this class of molecule.
Listed below are examples of co-factors and prosthetic groups.
Compound Function
Nicotinamide adenine dinucleotide (NAD+) hydrogen transfer
Flavin mono nucleotide hydrogen transfer
Flavin adenine dinucleotide (FAD) hydrogen transfer
Heme electron transfer
Ferredoxins electron transfer, hydrogen activation
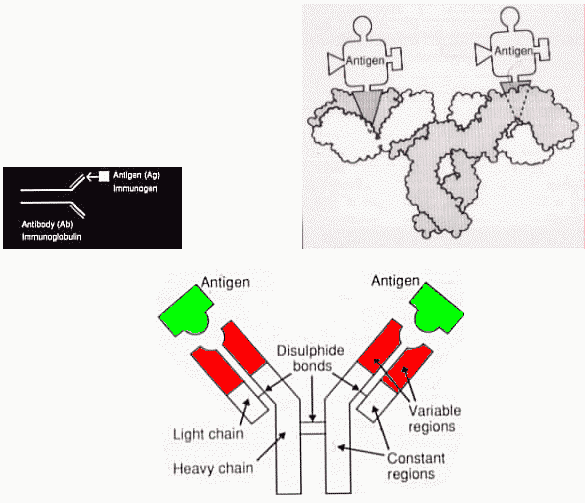
They are of one defined specificity.
They can be produced indefinately by cells in culture.
Large quantities of antibody can be obtained from hybridomas grown in mice.
The immunogen need not be pure.
They cut down on the number of lab animals required.
The alternative to using MAbs is using polyclonal antisera, however this had a number of disadvantages:
It is a mixture of antibodies of high and low affinity antibody populations.
It is a mixture of antibodies of different specificities.
The supply is limited to the life span of the immunized animal.
The quantity that is produced is small.
The immunogen has to be pure.
This limits the use of polyclonal antisera and advocates the use of MAbs.
Production of MAbs
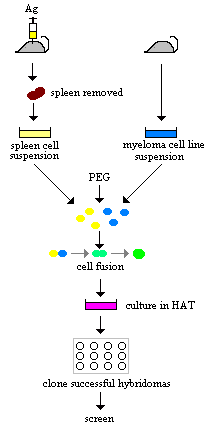
The generation of B-cell hybridomas by fusing antigen-primed B cells and myeloma cells.
Selection of the fused clones.
Screening of clones for antibody secretion with desired antigenic specificity.
Propagation of desired hybridomas.
Production of Human MAbs
Chimeric and hybrid antibodies.
Phage display antibodies.
Chimeric and hybrid MAbs
Immunoassay and Immunodetection
Immunocytochemistry
Immunoprecipitation
Immunoblotting
Immunoaffinity Purification
Immunoassay
highly sensitive
have low non-specific binding
able to assay a large number of samples in a short time
Immunocytochemistry
Cell or tissue preparation
Fixation and preparation of thinly sliced sections on slides
Antibody binding
Detection
Immunoprecipitation
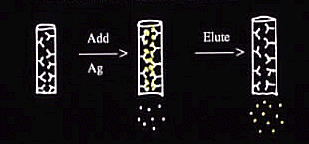
Research
Diagnostics
Therapeutics
Research
Blood and tissue typing - important in blood transfusions and transplantation.
Quantification of T cell levels in HIV patients
A screening test for prostate cancer - The PSA test
Diagnosis of microbial infections - it identifies the organism involved
Pregnancy testing - MAbs detect the human chorionic gonadotrophin hormone
A screening test for HIV.
Therapeutics
|
One of the most common pieces of equipment used to separate materials into subfractions in a biochemistry lab is the centrifuge. A centrifuge is a device that spins liquid samples at high speeds and thus creates a strong centripetal force causing the denser materials to travel towards the bottom of the centrifuge tube more rapidly than they would under the force of normal gravity. Types of centrifuges. The major distinguishing features between centrifuge types are speed and capacity. In a typical biochemistry laboratory you will find three different centrifuges (this is true in your biochemistry teaching lab as well). The smallest are the so-called microfuge centrifuges. These are made for spinning 1 to 2 ml plastic centrifuge tubes at speeds up to 12 or 13 thousand rounds per minute. They have very small, light rotors in them (the rotor is the part of the centrifuge that contains the holes for the sample tubes) which speed up and slow down rapidly. These centrifuges are very convenient for low to medium speed centrifugation of small quantities of material. The next common size centrifuge is the large superspeed centrifuge. These have speeds up to about 20,000 rpm and can take tubes of various sizes, depending on the rotors (the larger the rotor, the slower the maximum speed). Typical tubes hold 25 or 30 mls but bottles as large as several hundred mls can be run with the correct rotor. Finally, most biochemistry laboratories have access to an ultracentrifuge. Speeds up to 70,000 rpm are available on typical modern versions. Again the size of tube and the maximum speed vary from rotor to rotor, but tube sizes up to about 60 mls are available. The theory behind centrifugation. The idea here is pretty straight forward and mechanical. If you want the more dense materials to be separated from the less dense materials, you need a force that differentiates between particles of different density. Think about a swimming pool with a rock and a piece of styrofoam. The rock is denser than water and thus it sinks. The styrofoam is less dense than water, and thus it floats. Density is of course mass per unit volume. So, if you have a bag full of rocks and styrofoam and you want to separate one from the other, just dump the mixture into some water under the influence of the earth's gravity. The rocks will displace the water because they have greater mass for a given volume and gravity will pull them through the water. On the other hand, the water will displace the styrofoam because a certain volume of water weighs more than the same volume of styrofoam. However, there are many things that are much closer in density than rocks and styrofoam and it is much harder to separate them just under the Earth's gravity. In addition, diffusion is always at work as random motion smears out small differences due to density. To overcome this, or sometimes just to make the separation process faster, it would be nice to come up with a way of generating larger mass (density) dependent forces than are available from the Earth's gravity alone. Another way to generate a mass dependent force is to spin something. As you know from physics, a body in motion tends to continue in motion along a straight path unless some force is exerted on it to change its path. Thus in order to force something to go in a circle, we must exert force on it pulling it in towards the center. An equal and opposite force will always result, pushing out from the center. This is cetripital force, and it is just the mass of the object times the acceleration required to keep it from flying outward along a straight line. Thus, things with larger mass (for a given volume) will have a greater force exerted on them and they will move towards the outer edge of the container more quickly than the things with a lower mass per volume. The acceleration required to keep the object from flying outward along a straight path is given by w2r where the greek letter omega stands for the speed of revolution (see below for units) and r is the distance from the axis of the revolution to the position of the sample. To get a the force felt by the sample, we multiply this by the mass of the sample: F = mw2r, where F is the force and m is the mass. Please notice two things about this equation. It is linearly proportional to the mass of the object, but the force increases with the square of the rotation speed. Thus, going from 1000 rpm to 10,000 rpm increases the forces involved by a factor of 100. Some practical considerations. So let's say I want to spin two 30 mls tubes of water at 50,000 rpm. What kinds of forces are involved? Well, for comparison, consider the force that the acceleration of gravity exerts on the 30 mls of water when it is sitting on a bench top. The acceleration of gravity is about 10 m/s2 (10 meters per second squared). The mass of 30 mls of water is about 30 g or about 0.03 kg. Thus the force involved at 1 g (1 times the acceleration of gravity) is 0.3 kg m/s2 or 0.3 Newtons. How about at 50,000 rpm? First we need to do some units conversion. 50,000 rpm is 50,000/60 = 830 rounds per second. Further, in order to make the units work out, we must convert rounds per second to radians per second (there are 2p radians in a complete round or circle so multiply by this factor). This gives 5200 radians per second (this is omega in the equation). The force is mw2r so the total force is 66,000 N (assuming that the sample is about 8 cm from the center of rotation). To get the number of times greater this is than gravity along, we divide by 0.3 (see above) and get about 220,000 g's. That means that the water sample spinning at 50,000 rpm is equivalent to a 13,000 lb truck at normal gravity. If you look inside an ultracentrifuge, you will find that the rotor (the thing that contains the samples) is sitting on a shaft. The shaft is rather small and actually wobbly. There is no way you could suspend a 13,000 lb truck from this shaft. So how does it work? It works because of symmetry. You always must have a sample directly across from the sample of interest that has the same overall mass. Thus, the two masses and the forces on them balance out and the shaft feels no torque. The moral of the story is that you must balance tubes for centrifuge runs. For ultracentrifuge runs, you must balance the tubes very, very carefully. I usually try to get the masses within about 0.03 grams for an ultracentrifuge run. Slower centrifuges you can use an accordingly less stringent balance procedure. Rotors. There are many types of rotors, but most fall into one of three classes: fixed angle rotors, swinging bucket rotors and vertical rotors. Most of the time you will used a fixed angle rotor. Occasionally a swinging bucket or vertical rotor with be used, but not very often any form (swinging bucket allows the sample to swing out to the horizontal, giving the longest possible path of movement of the particles. The vertical rotors do the opposite -- they are used when a very short overall path of centrifugation is required. Types of centrifugation applications. There are many ways in which centrifuges are used. More often than not they are used to sediment some material leaving the rest in solution. However, one can also use two other common applications for separating materials: equilibrium density sedimentation and kinetic density sedimentation. In the first case, the material is either layered on top of or mixed into some material that can either be preformed into a density gradient or will become a density gradient when it is spun at high speed. The centrifuge is then run until the material finds its place as a band of particular density within the tube. The kinetic density methods also generally involve long runs that allow the molecule to find a region of the medium with the same density and come to equilibrium. In kinetic density sedimentation, you do not run the gradient to the end. You start with a band of your sample on top of the tube and let it progress through the density gradient for some period of time. |
Enzyme Kinetics; Cardosi, Marco, Electrochemical Sensors Group; 2001; University of Paisley, Paisley, Scotland, U.K.
Enzyme Kinetics, Natural Toxins Research Center; 2001; Texas A&M University, Kingsville, TX
Flow Cytometry; Martz, E., Cote, C.; 2000; The University of Massachusetts, Amherst, MA
Optical Microscopy (Molecular Expressions Review), Davidson, M.W., Abramowitz, M.; 1999; Florida State University, Tallahassee, FL
The Beer-Lambert Law; The Squirer Group; 1999; The University of California, San Diego, CA
|
|
|
| Search THIS ENTIRE WEB SITE for CLS technical, financial or other data >>> |
 E-mail
Irving E-mail
Irving |
This page is made entirely from recycled electrons. Any similarity between this and any other page may be completely intentional. Not tested on animals. Void where prohibited. Return to Irving's Home Page |